Peer Reviewed Proof
- E. Volk, E. Padgett, M. E. Kreider, S. Kwon, S. Alia, (2026) Voltage breakdown analyses in anion exchange membrane water electrolysis - the contributions of catalyst layer resistance on overall overpotentials, Energy Advances.
Abstract
Abstract
Despite many recent advances, overpotentials remain high for anion exchange membrane water electrolyzers (AEMWEs). Voltage breakdown analyses (VBA) can help decouple the origins of overpotentials and facilitate design decisions to improve cell performance, but studies investigating how to adapt and apply VBA to AEMWEs are lacking. Specifically, catalyst layer resistances and their contributions to overpotentials are not consistently quantified in water electrolysis and are rarely quantified for AEMWEs. This work presents a systematic methodology for VBA tailored to AEMWEs, including an approach to Tafel analysis in the absence of a reference electrode and under conditions where both the oxygen evolution reaction and hydrogen evolution reaction exhibit significant overpotentials. Catalyst layer resistance contributions are diagnosed via changes in the catalyst layer thickness, transport layer porosity, ionomer content, and electrolyte concentration. In this study, we explain discrepancies between inherent catalytic kinetics and device level performance and identify catalyst layer design strategies to reduce catalyst layer resistances.
- M. Vyas, K. Ithisuphalap, L. Patterson, M. J. Rasmussen, Y. Bian, W. Pham, S. Kwon, (2025) Confinement of Lewis Acid–Base Sites by Microporous Silica Layers on Titania for Enhanced Alkanol Dehydration Reactivity, ACS Catal., 15, 21019 - 21032.
Abstract
Abstract
Alkanol dehydration offers a pathway to upgrade biomass-derived short-chain oxygenates into alkenes, essential chemical building blocks widely used in industrial applications. Transition metal oxides with Lewis acid–base site pairs are attractive catalysts due to their high reactivity and cost-effectiveness. This work demonstrates a synthetic pathway to manipulate local environments around active Lewis acid–base pairs in anatase TiO2 to enhance their reactivity in alkanol dehydration. Microporous SiO2 layers with an average pore diameter of ∼0.6 nm and a controlled thickness of 0.8–33 nm are deposited on anatase TiO2 powders by using a molecular templated SiO2 deposition method. The Lewis acid–base strength of accessible Ti–O pairs remains unchanged, as shown by temperature-programmed surface reactions of surface-bound formic acid-derived species and temperature-programmed desorption of pyridine. However, measured alkanol dehydration rates on confined Ti–O pairs are much higher (by up to 7-fold) than those on TiO2. The extent of rate enhancements depends on the reactant size and functional group positioning, suggesting that the rate enhancements reflect the interactions between the guest molecules (reactants and transition states) and the surrounding SiO2 micropore environments. By providing a detailed synthetic procedure to tailor the local environments around active sites in bulk oxides, this approach offers an additional avenue for enhancing catalytic performance.
- M. Vyas, L. Patterson, S. Kwon, (2025) Alkanol dehydration and dehydrogenation pathways on anatase TiO2 under oxidative and nonoxidative conditions, J. Cat., 452, 116414.
Abstract
Highlights
- 2-Propanol reactions on anatase TiO2 form propene and acetone.
- Propene forms via dehydration on Lewis acid base Ti–O sites.
- Without O2, Acetone forms via non-oxidative dehydrogenation at O-vacancy sites.
- With O2 co-feed, acetone forms via oxidative dehydrogenation on lattice O-atoms.
Abstract
Alkanol dehydration offers a promising route for converting biomass-derived short-chain oxygenates into alkenes, which are valuable industrial intermediates. Titanium dioxide (TiO2) has been extensively studied for this reaction due to its high activity and low cost. However, the inherent heterogeneity of commercially available TiO2 has led to conflicting reports on reactivity and product selectivity, further complicated by the diverse reaction conditions employed in previous studies. This work aims to update our understanding of alkanol reactions on anatase TiO2 by integrating transient, titration, kinetic, and spectroscopic methods, using 2-propanol and 1-propanol as model reactants. Under anerobic conditions, 2-propanol primarily undergoes dehydration to form propene and water. A minor pathway forms acetone via nonoxidative dehydrogenation, but only after an induction period during which surface oxygen vacancies accumulate. Notably, propene formation rates remain largely constant, even as acetone formation rates decrease by ∼60 % during the initial induction period. These transient behaviors, together with pretreatment and characterization data, suggest that dehydration predominantly occurs on smaller TiO2 crystallites that provide most of the surface area, whereas dehydrogenation is favored on larger crystallites with smaller band gap energies. In situ pyridine titration was used to quantify the number of active sites for each pathway, enabling accurate turnover rate normalization. Detailed kinetic analysis showed that both dehydration and nonoxidative dehydrogenation are inhibited by 2-propanol, water, and 2-propanol–water dimers, but to different extents. The rate constant for nonoxidative dehydrogenation reflects a significantly lower activation enthalpy than that for the oxidative route; however, this is offset by a large entropic penalty, resulting in higher free energy barriers at relevant temperatures and thus a lower overall rate constant. This kinetic framework offers mechanistic insight into the temperature- and pressure-dependent shifts in reaction rates and product selectivity, while also reconciling prior discrepancies reported in the literature.
- E. Volk, M. E. Kreider, D. Gibson Colón, M. Müller, S. Sunde, S. Alia, S. Kwon, (2025) Electrochemical Activation of Ni–Fe Oxides for the Oxygen Evolution Reaction in Alkaline Media, ACS Catal., 15, 11475 - 11486.
Abstract
Abstract
The oxygen evolution reaction (OER) is essential to many key electrochemical devices, including H2O electrolyzers, CO2 electrolyzers, and metal–air batteries. NiFe oxides have been historically identified as active for the OER, though they have been less studied in their more commercially relevant bulk oxide forms, such as NiFe2O4. Past works have demonstrated that the initial starting phase of Ni(Fe) precatalysts can influence their activation to the Ni(Fe)OOH active phase, including the rate and degree of conversion, pointing to the necessity of understanding activation protocols and in situ characteristics of catalyst materials at the device level. In this work, we investigate the characteristics of commercially relevant NiFe bulk oxides (NiFe2O4 and a physical mixture of NiO and γ-Fe2O3) during multiple activation procedures. Our results demonstrate that significant performance enhancement is observed for these bulk oxides regardless of the Fe incorporation in the initial form (i.e., atomically or macroscopically integrated), leading to significant performance enhancement (up to 30×) over time on stream. We hypothesize that this activation is due to the formation of NiFeOOH active sites on the surface, supported by in situ cyclic voltammetry and Raman spectroscopy results. We further show that not only the starting material but also the method of activation influences the number of Ni(Fe)OOH active sites formed and suggest that these sites can be quantified from the Ni2+ to Ni3+ redox transition using cyclic voltammetry. Broadly, this work demonstrates the necessity of in situ characterization of catalyst materials for cell-level design and testing.
- T. C. Lin, M. Nolen, C. Farberow, S. Kwon, A. Bhan (2025) Mechanistic and kinetic relevance of hydrogen and water in CO2 hydrogenation on Cu-based catalysts, J. Cat., 433, 115936.
Abstract
Highlights
- Cu/ZnO/Al2O3 and Cu/Al2O3 exhibit identical reaction orders for CO2 hydrogenation.
- Methanol synthesis and reverse water-gas shift occur on a common active site.
- Active sites are saturated by H* and HCOOH** surface species during catalysis.
- Competing pathways of CO2 hydrogenation involve distinct rate-determining steps.
- H2O preferentially inhibits methanol synthesis and alters its rate-determining step.
Abstract
We ally steady-state kinetics, kinetic isotope effects, and density functional theory (DFT) calculations to illustrate that Cu-based catalysts remain saturated by H-adatoms (H*) and molecular formic acid (HCOOH**) during CO2 hydrogenation. High H* coverage under methanol synthesis conditions is evidenced by reverse water-gas shift (RWGS) rates that exhibit positive H2 reaction orders only at PH2 < 0.5 bar, above which methanol synthesis and RWGS rates exhibit first and zeroth order dependence on PH2, respectively. HCOOH** also accumulates on the surface with increasing PCO2 as informed by the Langmuir-type dependence on PCO2 (0.25-23 bar) for both methanol synthesis and RWGS. As both HCOOH** and H* have one H-atom per site occupied, the two species share the same PH2 dependence and give rise to CO2 reaction orders that are independent of PH2. Surface coverages determined based on kinetic analyses are further corroborated with DFT-derived adsorption energies that show favorable HCOOH** adsorbate-adsorbate interactions as well as repulsive interactions for bidentate formate (HCOO**) on H*-saturated surfaces. Methanol selectivity remains invariant with PCO2 and PCO despite CO inhibiting reaction rates, thereby demonstrating methanol synthesis and RWGS occur on the same active site. In contrast, water preferentially inhibits methanol synthesis rates, increases methanol synthesis H2 reaction order from 1.0 to 1.5, and alters the methanol synthesis H2/D2 kinetic isotope effect; the inhibitory effect of H2O thus cannot be attributed to competitive adsorption alone and instead reflects a change in the rate-determining step for methanol synthesis. The disparate kinetics of methanol synthesis and RWGS evince a branching pathway where methanol is formed from formates and CO is formed from carboxylates. The presented work thus identifies the relevant surface species, underscores the distinct catalytic role of water in branching methanol synthesis and RWGS pathways, and, in doing so, details a mechanistic picture that yields predictable rates and reaction orders for both methanol synthesis and RWGS on Cu-based CO2 hydrogenation catalysts.
- E. Volk, A. Clauser, M. E. Kreider, D. Soetrisno, S. Khandavalli, J. Sugar, S. Kwon, S. Alia, (2024) The role of the ionomer in supporting electrolyte-fed anion exchange membrane water electrolyzers, ACS Electrochem., 1(2), 239 - 248.
Abstract
Abstract
While anion exchange membrane water electrolyzers (AEMWEs) have achieved significant performance advances in recent decades, overpotentials remain high relative to their proton exchange membrane water electrolyzer (PEMWE) counterparts, requiring AEMWE-specific catalyst layer design strategies to further advance this technology. In this work, the role of the ionomer in catalyst layer structure and quality, catalyst layer stability, and ion conduction for supporting electrolyte-fed AEMWEs is assessed for catalyst layers composed of NiFe2O4 and PiperION TP85 from Versogen at variable ionomer contents (0-30 wt %) for tests up to 200 h. The results reveal that, for supporting electrolyte-fed AEM devices, the ionomer is not required for ion conduction through the catalyst layer. Instead, the ionomer is found to play a critical role in catalyst layer structure and stability, where intermediate ionomer contents lead to the lowest overpotentials, highest effective surface areas, and lowest catalyst layer resistances. Catalyst layer stability is found to be a function of both catalyst adhesion and ionomer loss. These results show that an ionomer may be selected which is not of the same chemistry as the anion exchange membrane, mitigating ionomer stability concerns throughout the catalyst layer and offering a pathway towards highly active and stable AEMWEs.
- M. Nolen, C. Farberow, S. Kwon (2024) Incorporating Coverage-Dependent Reaction Barriers into First-Principles-Based Microkinetic Models: Approaches and Challenges, ACS Catal., 14(18), 14206 - 14218.
Abstract and key graphics
Abstract
Mean-field microkinetic models (MKMs) are appealing for their relatively facile construction, computational tractability, and high-throughput catalyst screening capabilities. As such, they will continue to be a valuable tool for materials design in heterogeneous catalysis even as the field aims to describe more complex systems. Numerous prior reports have provided the groundwork for constructing first-principles-based MKMs, including the analysis of strategies for incorporating lateral interactions into thermodynamic parameters (e.g., adsorption energies). Yet, there remains a need for concerted dialogue on methods for calculating and incorporating coverage-dependent kinetic parameters into MKMs. In this Perspective, we assess strategies for doing so, including the corresponding key physical implications and computational challenges. We emphasize that decoupling thermodynamic and kinetic parameters within MKMs can violate thermodynamic consistency and risk unphysical solutions. For some reactions and catalyst materials, scaling relationships can predict coverage-dependent activation energies, but there are several exceptions evident in the literature, indicating that this approach is not universally applicable and that the field could benefit from research aimed at elucidating the limitations. Conducting high-coverage transition state searches is a rigorous but computationally costly strategy, and the effects of various methods for mitigating this cost on resulting energetics have yet to be broadly explored and validated. The goal of this Perspective is to generate discussion on and inspire focused research into the physical relevance of approaches for describing coverage-dependent reaction barriers in MKMs, including the development of computationally tractable methodologies, to advance the applicability of MKMs across diverse reaction chemistries and conditions.
- C. J. Wrasman, A. T. Bell, B. D. Chandler, J. W. Harris, S. Kwon, M. R. Ball, … & J. A. Schaidle (2024) Recommendations for improving rigor and reproducibility in site specific characterization, J. Cat., 433, 115451.
Abstract and key graphics
Abstract
Heterogeneous catalysis is driven by the interaction of reactant molecules and the catalyst surface. The locus of this interaction as well as the surrounding ensemble of atoms is referred to as the catalyst active site. Active site characterization attempts to distinguish active catalytic sites from inactive surface sites, to elucidate the structural and chemical nature of active sites, and to quantify active site concentration. Numerous techniques have been demonstrated to provide compositional and structural information about the active sites within a catalyst. However, each technique has its own limitations and experimental pitfalls that can lead to data misinterpretation or irreproducible results. This work aims to provide an overview of the types of data that can be collected, to outline common experimental challenges and how to avoid them, and to assemble relevant references for the most used active site characterization techniques. More broadly, we aim to outline best practices for researchers to collect, interpret, and report active site characterization data in a way that provides the most benefit to the broader catalysis community. Increasing the rigor and reproducibility of active site characterization offers a strategy to better link properties with catalytic performance and to enable the community to develop consensus concerning these relationships.
- K. Ithisuphalap, M. Nolen, H. Monroe, S. Kwon (2023) Kinetic, spectroscopic, and theoretical study of toluene alkylation with ethylene on acidic Mordenite zeolite, ACS Catal., 13, 16012 - 16031.
Abstract and key graphics
Abstract
This work combines kinetic, spectroscopic, and theoretical methods to understand mechanistic details of toluene alkylation with ethylene on acidic MOR zeolite, chosen due to its industrial relevance in producing ethyltoluene. In doing so, we show that the protons in the small eight-membered-ring (8-MR) micropores are inaccessible to large toluene molecules, which were selectively titrated with Na+ prior to kinetic measurements. Kinetic data, taken together with in situ infrared spectra and density functional theory (DFT) calculations, show that all protons in the 12-MR are saturated with π-bonded toluene, favoring thermodynamic stability due to extensive dispersive interactions with the porous structure. The π-bonded toluene reacts with ethylene in a concerted manner, where the protonation of ethylene and C–C coupling occurs concurrently. The C–C formation at the *ortho* position takes precedence over *meta* and *para* locations, although the free energies of C–C coupling transition states show marginal differences. Consequently, all three isomers are formed as primary products, with an *ortho*, *meta*, and *para* ratio of 3:1:1 (<0.2% conversion; 503 K). Undesired ethylene dimerization products remained undetectable on partially Na+-exchanged MOR samples under all conditions, consistent with larger DFT-derived barriers for C–C coupling between two ethylene molecules than between toluene and ethylene, reflecting the role of 12-MR micropores that selectively stabilize the latter transition state. These results provide mechanistic insights into aromatic alkylation and the role of micropores on rates and selectivities, which will ultimately inform design strategies for solid acid catalysts with improved catalytic performance.
- E. Volk, M. E. Kreider, S. Kwon and S. Alia. (2023) Recent progress in understanding the catalyst layer in anion exchange membrane electrolyzers – durability, utilization, and integration, EES Catalysis, 2, 109 - 137.
Abstract and key graphics
Abstract
Anion exchange membrane water electrolyzers (AEMWEs) are poised to play a key role in reducing capital cost and materials criticality concerns associated with traditional low-temperature electrolysis technologies. To accelerate the development and deployment of this technology, an in-depth understanding of cell materials integration is essential. Notably, the complex chemistries and interactions within the catalyst layer (consisting of the anode/cathode catalyst, anion exchange ionomer, and their interfaces with the transport layers and membrane) collectively influence overall cell performances, lifetimes, and costs. This review outlines recent advances in understanding the catalyst layer in AEMWEs. Specifically, electrode development strategies (including catalyst deposition techniques and configurations as well as transport layer design strategies) and our current understanding of catalyst–ionomer interactions are discussed. Effects of cell assembly and operational variables (including compression, temperature, pressure, and electrolyte conditions) on cell performance are also discussed. Lastly, we consider cutting-edge *in situ* and *ex situ* diagnostic techniques to study the complex chemistries within the catalyst layer as well as discuss degradation mechanisms that arise due to the integration of cell components. Simultaneously, comparisons are made to proton exchange membrane water electrolyzers (PEMWEs) and liquid alkaline water electrolyzers (LAWE) throughout the review to provide context to researchers transitioning into the AEMWE space. We also include recommendations for standard operating procedures, configurations, and metrics for comparing activity and stability.
- M. Nolen, S. A. Tacey, S. Kwon, and C. Farberow. (2023) Theoretical assessments of CO2 activation and hydrogenation pathways on transition-metal surfaces, Appl. Surf. Sci, 637, 157873.
Abstract and key graphics
Abstract
Carbon dioxide (CO2) hydrogenation on transition-metal active sites offers a promising carbon utilization route toward mitigating greenhouse gas emissions. C1 products are often formed in parallel during CO2 hydrogenation, prompting investigations into the intrinsic properties of transition metals that drive activity and product selectivity. In this work, close-packed surfaces of a selection of transition-metal catalysts (Ni, Co, Rh, Ru, Pd, and Pt) were studied with density functional theory (DFT) calculations to understand their fundamental reactivities for CO2 transformation reactions. Results indicate that CO2 conversion proceeds through CO* formation and hydrogenation to form C1 products (* denotes an adsorbed species). Ni, Co, Rh, and Ru favor CO/CH4 formation, while Pd and Pt favor CO/CH3OH formation. The ability of a metal to dissociate C-O bonds drives selectivity between CH4 and CH3OH, while competition between CO* desorption and surface hydrogenation describes CO selectivities. The C-O bond dissociation steps often impose the highest barrier along CH4 formation reaction profiles, suggesting their kinetic relevance for CH4 formation rates. The provided DFT-derived data sets detail a comprehensive reaction network of elementary steps relevant to C1 chemistries, ultimately offering a benchmark for insights into design strategies for materials that exploit transition-metal active sites in carbon capture or utilization processes.
- M. Vyas, F. Fajardo-Rojas, D. A. Gómez-Gualdrón, and S. Kwon. (2023) Theoretical assessments of Pd-PdO phase transformation and its impacts on H2O2 synthesis and decomposition pathways, Catal. Sci. Technol., 13, 3828 - 3848. (Emerging Investigator Series).
Abstract and key graphics
Abstract
The direct synthesis of H2O2 from O2 and H2 provides a green pathway to produce H2O2, a popular industrial oxidant. Here, we theoretically investigate the effects of Pd oxidation states, coordination environments, and particle sizes on primary H2O2 selectivities, assessed by calculating the ratio of rate constants for the formation of H2O2 (*via* OOH* reduction; kO–H) and the decomposition of OOH* (via O–O cleavage; kO–O). For Pd metals, the kO–H/kO–O ratio decreased from 10−4 for Pd(111) to 10−10 for the Pd13 cluster at 300 K, indicating poorer H2O2 selectivity as Pd particle size decreases and low primary selectivities for H2O2 overall. As the oxygen chemical potential increases and metals form surface and bulk oxides, the perturbation of Pd–Pd ensemble sites by lattice O atoms results in selectivities that become dramatically higher than unity. For instance, at 300 K, the kO–H/kO–O ratio increases significantly from 10−4 to 109 to 1016 as Pd(111) oxidizes to Pd5O4/Pd(111) and to PdO(100), respectively. In contrast, such selectivity enhancements are not observed for surface and bulk oxides that persistently contain rows of more metallic, undercoordinated Pd–Pd ensemble sites, such as PdO(101)/Pd(100) and PdO(101). These Pd–Pd ensembles are also absent when smaller Pd nanoparticles fully oxidize, indicating that smaller PdO clusters can be more selective for H2O2 synthesis. These trends for primary H2O2 selectivities were found to inversely correlate with trends for H2O2 decomposition rates via O–O bond cleavage, demonstrating that catalysts with high primary H2O2 selectivity can also hinder H2O2 decomposition. Ab initio thermodynamic calculations are used to estimate the thermodynamically favored phase among Pd, PdO/Pd and PdO in O2, H2O2/H2O, and O2/H2 environments. These results are combined to show that smaller Pd nanoparticles are more prone to be oxidized at lower oxygen chemical potentials, upon which they become more selective than larger Pd particles for H2O2 synthesis.
- E. Volk, S. Kwon and S. Alia. (2023) Catalytic activity and stability of non-platinum group metal oxides for the oxygen evolution reaction in anion exchange membrane electrolyzers, J. Electrochem. Soc., 170(6), 064506.
Abstract and key graphics
Abstract
The activities and stabilities of non-platinum group metals (PGMs) in the forms of monometallic (Mn2O3, Fe2O3, Co3O4, NiO) and bimetallic (NiFe2O4, CoNiO2) oxides were assessed for the oxygen evolution reaction (OER) in alkaline media and compared with IrO2. Both half-cell, rotating disc electrode (RDE) apparatus and single-cell, membrane electrode assemblies (MEA) were used to study kinetic and device-level performance in parallel and to provide insights into the use of these materials in anion exchange membrane (AEM) electrolyzers. Normalization of RDE results by geometric and physical surface areas, double layer capacitance, and metal content probed differences in physically vs electrochemically accessible surface areas and ensured reported trends were independent of the normalization method. The results showed that: (i) Ni- and Co- containing materials met or exceeded IrO2 performance in both RDE and MEA testing, (ii) Co3O4 deactivated over time-on-stream (1.8 V for 13.5 h) due to oxide and, relatedly, particle growth, (iii) NiFe2O4 increased in activity over time-on-stream due to dissolution of Fe and an increased Ni/Fe ratio, and (iv) reduction of catalyst layer resistance is an avenue to further increase device-level performance. These results demonstrated the clear viability for non-PGMs to be used as anode catalysts in AEM devices.
- S. Schlussel and S. Kwon. (2022) (Invited Review Paper) A review of formic acid decomposition routes on transition metals for its potential use as a liquid H2 carrier, Korean J. Chem. Eng., 39(11), 2883 - 2895.
Abstract and key graphics
Abstract
Formic acid (HCOOH) has emerged as a promising H2 energy carrier due to its reasonable gravimetric and volumetric H2 densities, low toxicity, low flammability, and ease of handling. Its possible productions from biogenic feedstocks also make it an attractive source to produce H2 on demand. The utilization of HCOOH as a liquid H2 carrier requires catalytic systems to selectively dehydrogenate HCOOH at low temperatures without forming CO products that can act as a poison in fuel cell applications. In this review, we summarize the recent progress in understanding HCOOH decomposition via dehydrogenation (to CO2/H2) and dehydration (to CO/H2O) pathways on transition metals, including Cu, Pt, Pd, and Au. The focus is on discussing the surface chemistry of HCOOH reactions on transition metals, including the types of bound intermediates and the identity and kinetic relevance of elementary steps. In doing so, we review current catalyst design strategies for HCOOH dehydrogenation to facilitate the future development of catalytic processes for H2 storage/utilization.
- T. C. Lin, U. De La Torrea, A. Hejazi, S. Kwon, and E. Iglesia. (2021) Unimolecular and bimolecular formic acid decomposition routes on dispersed Cu nanoparticles, J. Cat., 404, 814 - 831.
Abstract and key graphics
Key graphics
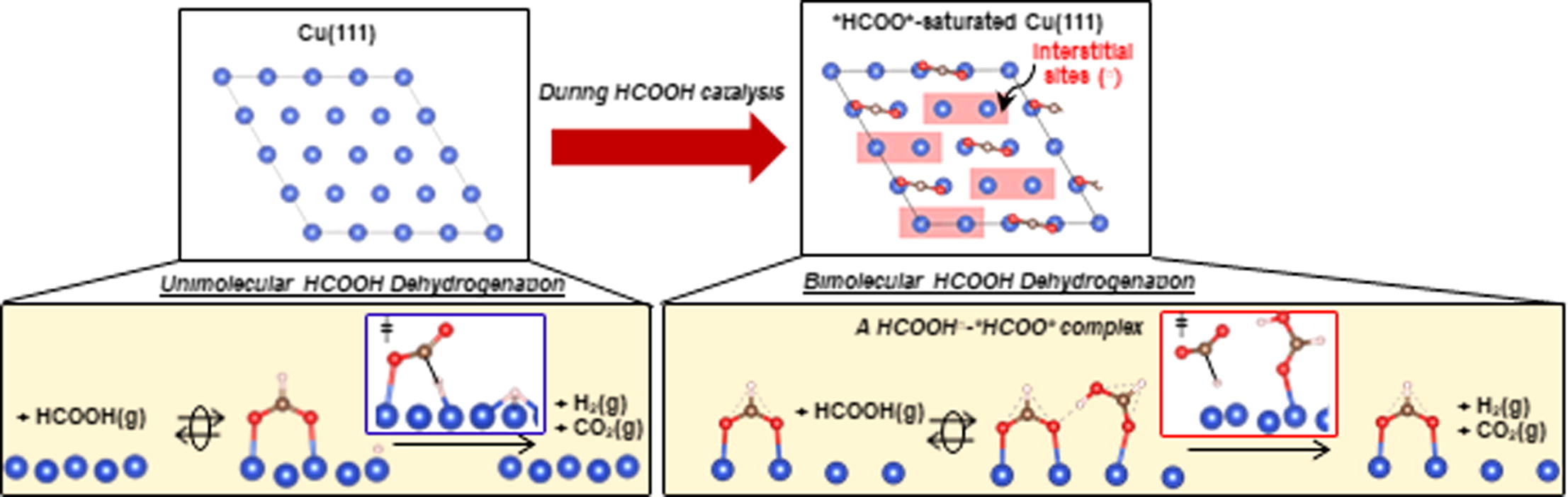
Highlights
- Cu surfaces are saturated with bidentate formates (*HCOO*) during catalysis.
- *HCOO* species saturate at 0.25 monolayer (0.25 *HCOO* per Cu surface atom).
- HCOOH adsorbs molecularly at interstices (□) within the *HCOO* adlayer.
- The coexisting HCOOH□ enables *HCOO* to decompose bimolecularly.
- Biomolecular decomposition occurs at lower barriers than the unimolecular route.
Abstract
The elementary steps and site requirements in formic acid (HCOOH) dehydrogenation on Cu surfaces remain of keen interest because formate species act as intermediates or spectators in methanol synthesis and water–gas shift reactions. Steady-state and transient kinetic data, isotopic effects, infrared spectra during catalytic and stoichiometric reactions, and theoretical treatments based on density functional theory (DFT) provide evidence for bimolecular reactions, in which saturated bidentate formate (*HCOO*) adlayers, present at 0.25 ML (0.25 *HCOO* per surface Cu atom), react with undissociated species (HCOOH□) bound at interstices within formate adlayers (□) to form H-bonded bimolecular HCOOH□-*HCOO* adducts. The co-existence of vicinal HCOOH□ and *HCOO* moieties is evident from antisymmetric infrared bands for *HCOO* that become stronger as a result of their H-bonding that perturbs the induced dipole moment of *HCOO* upon vibration, consistent with DFT-derived vibrational frequencies and intensities for such perturbed species. The *HCOO* moiety in this complex undergoes C-H activation via a transition state that is preferentially stabilized through H-bonding with the vicinal HCOOH□ relative to its *HCOO* precursor. DFT-derived HCOOH dehydrogenation activation barriers and those determined from the evolution of CO2 from pre-adsorbed *HCOO* species are about 10 kJ mol−1 smaller in the presence of gaseous HCOOH reactants (because of HCOOH□-*HCOO* interactions) than those for the unimolecular decomposition of bound *HCOO* species. Such bimolecular routes are consistent with measured effects of HCOOH, H2, and CO pressures and of H/D isotopic substitution on dehydrogenation turnover rates and represent the predominant channel for the formation of CO2 and H2 during catalytic HCOOH dehydrogenation on Cu nanoparticles. A saturated *HCOO* adlayer that retains binding interstices and the presence of HCOOH(g) enable a sequence of elementary steps unavailable for *HCOO* species, thus circumventing unassisted unimolecular routes that exhibit higher activation barriers.
- S. Kwon, T. C. Lin, and E. Iglesia. (2020) Formic acid dehydration rates and elementary steps on Lewis acid-base site pairs at anatase and rutile TiO2 surfaces, J. Phys. Chem. C, 124(37), 20161 - 20174.
Abstract and key graphics
Key graphics
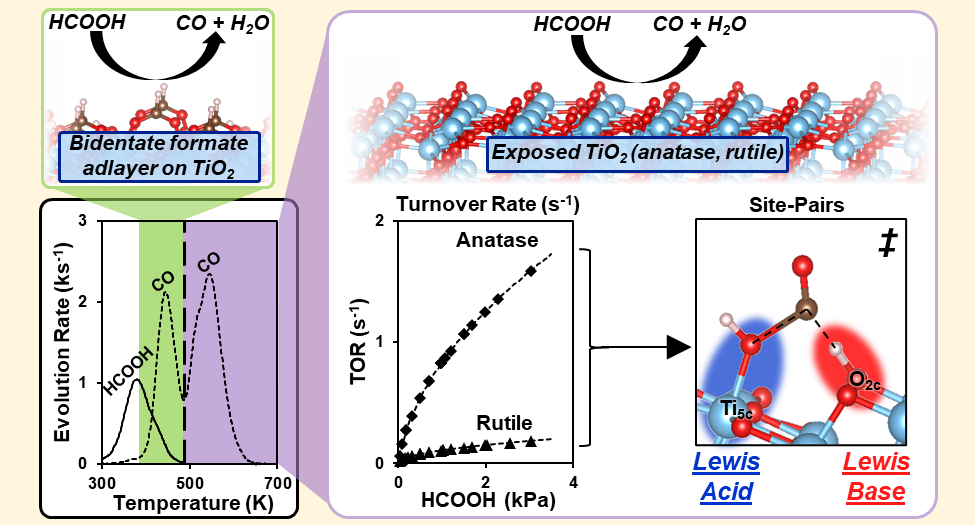
Abstract
Formic acid (HCOOH) decomposition is often used to assess the acid–base properties of oxide surfaces. Its reverse reaction forms HCOOH and formate species that can act as intermediates in CO2/CO/H2/H2O reactions that are important in C1 conversions. This study describes the mechanism of HCOOH dehydration on acid–base pairs at anatase and rutile TiO2 surfaces through spectroscopic, desorption-reaction, kinetic, isotopic, and theoretical methods. HCOOH dehydration turnover rates are measured at coverages that allow bound intermediates to interact directly with Ti5c–O2c pairs. Such rates then reflect their acid–base properties without interference from a refractory bidentate formate adlayer that acts as the catalytic surface at lower temperatures, as evident from infrared and desorption reaction data. HCOOH dehydration elementary steps involve the concurrent activation of C–O and C–H bonds in a molecularly bound HCOOH (HCOOH*) by a Ti5c–O2c pair at the kinetically relevant step. The transition state mediating this step involves the OH group and the H-atom of the C–H group in HCOOH* that are almost fully transferred to the Ti5c and the vicinal O2c center, respectively. Such concerted interactions with the acid and base centers and the late character of the transition state render the H2O dissociation energy at Ti5c–O2c pairs a more suitable descriptor of HCOOH reactivity than the respective strengths of each Lewis center. These mechanistic conclusions allow quantitative inferences of the rate and kinetic parameters for HCOOH synthesis from CO–H2O reactants on TiO2 surfaces through the tenets of microscopic reversibility extended to the sequence of elementary steps. The results also illustrate how acid–base pairs act in concert to stabilize the relevant transition states, thus making the balance between acid and base strengths, instead of their independent properties, the rigorous arbiters of reactivity, as shown by the similar reactivities and H2O dissociation energies on Ti5c–O2c pairs at anatase and rutile surfaces in spite of their very different acid and base strengths.
- Wu, W., S. Kwon, J. A. McCarthy, P. C. Stair, and E. Weitz (2020) Mechanistic Studies of the Oxidation of Cyclohexene to 2-Cyclohexen-1-one over ALD Prepared Titania Supported Vanadia, J. Phys. Chem. C, 124, 22, 11844 - 11862.
Abstract and key graphics
Key graphics
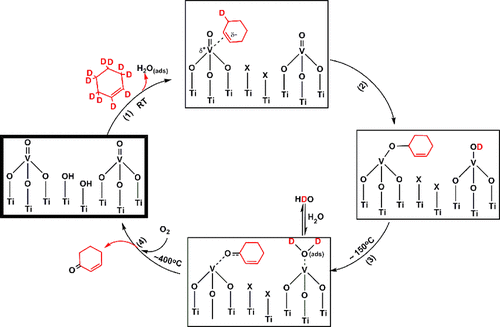
Abstract
Selective oxidation of cyclohexene to 2-cyclohexen-1-one over titania supported vanadia (VOx/TiO2) has been studied using temperature dependent in situ FTIR spectroscopy in both the presence and absence of oxygen. The VOx/TiO2 samples were prepared using one atomic layer deposition (ALD) cycle and characterized by Raman spectroscopy. In situ FTIR data for the oxidation of cyclohexene and perdeuterocyclohexene allow for the formulation of a molecular level reaction mechanism, which is initiated by the transfer of an allyl hydrogen. Oxidation of perdeuterocyclohexene provides a direct probe of the formation of OD and HDO moieties that support the involvement of specific steps in the proposed mechanism. The presence of gas phase oxygen does not lead to a change in the products versus anaerobic conditions. However, gas phase oxygen is significantly incorporated in the CO2 overoxidation product above ∼250 °C. Data were also obtained with cyclohexene epoxide as the reactant in an effort to determine whether there is a parallel reaction pathway, which is initiated by C═C activation in cyclohexene, that involves cyclohexene epoxide as an intermediate. Though a minor pathway involving a cyclohexene epoxide intermediate cannot be ruled out, these data demonstrate that, under experimental conditions, the dominant pathway from cyclohexene to cyclohexene-1-one is initiated by an allyl-H activation step and does not involve an epoxide intermediate.
- S. Kwon, T. C. Lin, and E. Iglesia. (2020) Elementary steps and site requirements in formic acid dehydration reactions on anatase rutile TiO2 surfaces, J. Cat., 383, 60 - 76.
Abstract and key graphics
Key graphics
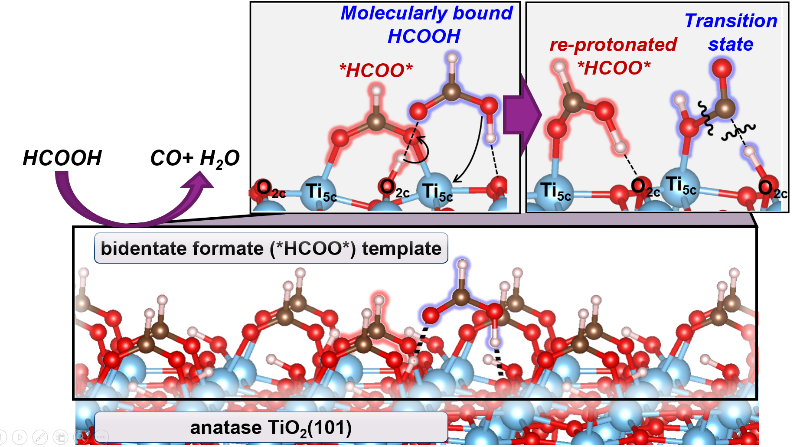
Highlights
- Decomposition of HCOOH on TiO2 surfaces forms CO and H2O products
- Inactive bidentate formates (*HCOO*) saturate all Ti5c centers at conditions of catalysis (423–463 K)
- HCOOH adsorbs molecularly at a proton (HCOOH-H*) in this *HCOO*-template
- HCOOH-H* eliminates H2O by reacting with a Ti5c-O2c pair, made available by the momentary reprotonation of *HCOO*
- The reprotonation of *HCOO* is less facile on rutile than on anatase TiO2, causing its lower reactivity at these conditions
Abstract
Mechanistic details of HCOOH decomposition routes provide valuable insights into reactions involving bound formates as intermediates or spectators; these routes are also widely used as a probe of the acid-base properties of oxide surfaces. The identity and kinetic relevance of bound intermediates, transition states, and elementary steps are reported here for HCOOH dehydration on anatase and rutile TiO2 surfaces through complementary kinetic, isotopic, spectroscopic and theoretical assessments. Five-coordinate exposed Ti5c centers are saturated with bidentate formates (*HCOO*) at catalytic conditions (423–463 K; 0.1–3 kPa HCOOH), as evident from infrared spectra collected during catalysis and the amounts of HCOOH and CO evolved upon heating the TiO2 samples containing pre-adsorbed HCOOH-derived species. These *HCOO* species are inactive but form a stable “surface template” that contains stochiometric protons onto which HCOOH binds molecularly (HCOOH-H*) to form a coexisting adlayer. H2O elimination from HCOOH-H* is the sole kinetically-relevant step. DFT-derived barriers show that this step involves its reaction with Ti5c-O2c that acts as a Lewis acid-base pair. Such route, in turn, requires the access of HCOOH-H* to a Ti5c center, which is made available through a momentary reprotonation of a *HCOO*. This step is much less facile on rutile than on anatase due to stronger acid strength of its Ti5c centers that binds *HCOO* species more strongly and its shorter Ti5c-Ti5c distances that induce greater repulsions between co-adsorbed HCOOH* formed upon reprotonation step. These differences account for low dehydration reactivity of rutile at these temperatures. This mechanistic interpretation is in full accord with DFT-derived barriers, binding energies, and kinetic isotope effects that quantitatively agree with the values from regressed kinetic and thermodynamic parameters, with in-situ infrared spectra that identify HCOOH-H* species as the sole reactive intermediates, and with the differences in turnover rates between anatase and rutile catalysts. These dehydration routes are also consistent with the surface chemistry expected for Lewis acid-base pairs on stoichiometry TiO2 surfaces without requiring the presence or involvement of reduced centers or titanols in the catalytic cycle. The reaction routes described in this work show how strongly-bound species, evident in presence and unreactive nature from in-situ infrared spectra, provide an organic “permanent” template for reactions of weakly-bound species that are often invisible in spectroscopy.
Before Colorado School of Mines
- S. Kwon, P. Deshlahra, and E. Iglesia. (2019) Reactivity and Selectivity Descriptors of Dioxygen Activation Routes on Metal Oxides, J. Cat., 377, 692 - 710.
Abstract and key graphics
Key graphics
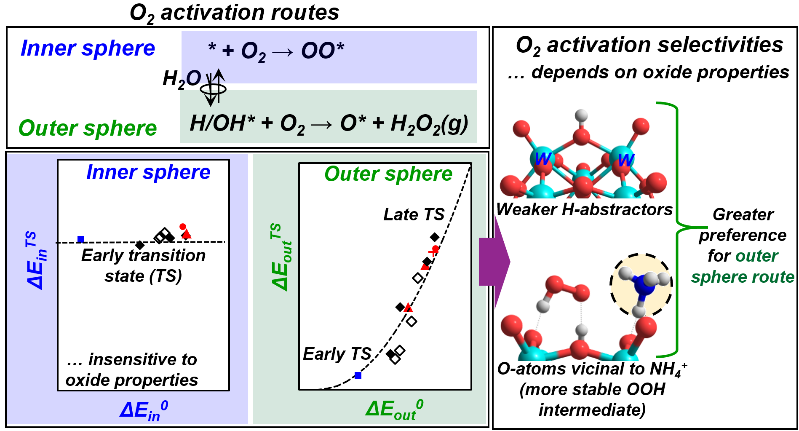
Highlights
- Two-electron reduced centers formed on metal oxides in redox cycles activate O2 via inner or outer sphere routes
- Inner sphere routes form bound peroxo species at O-vacancies
- Outer sphere routes form H2O2(g) at vicinal OH pairs, formed via H2O dissociation on O-vacancies
- The O-atoms in more reducible oxides exhibit a greater preference for the inner sphere routes
- The large charge-balancing cations influence the O2 activation selectivity of the vicinal O-atoms
Abstract
The activation of dioxygen at typically isolated two-electron reduced centers can lead to the formation of electrophilic superoxo or peroxo species, providing an essential route to form reactive O2-derived species in biological, organometallic, and heterogeneous catalysts. Alternatively, O2 activation can proceed via outer sphere routes, circumventing the formation of bound peroxo (OO*) species during oxidation catalysis by forming H2O2(g), which can react with another reduced center to form H2O. The electronic and binding properties of metal oxides that determine the relative rates of these activation routes are assessed here by systematic theoretical treatments using density functional theory (DFT). These methods are combined with conceptual frameworks based on thermochemical cycles and crossing potential models to assess the most appropriate descriptors for the activation barriers for each route using Keggin polyoxometalates as illustrative examples. In doing so, we show that inner sphere routes, which form OO* species via O2 activation on the O-vacancies (*) formed in the reduction part of redox cycles, are mediated by early transition states that only weakly sense the oxide binding properties. Outer sphere routes form H2O2(g) via O2 activation on OH pairs (H/OH*) formed by dissociation of H2O on O-vacancies; their rates and activation barriers reflect the rates of the first H-atom transfer from H/OH* to O2. The activation barriers for this H-transfer step depend on the binding energy of more weakly-bound H-atom in H/OH* pairs (HAE2) and on the .OOH-surface interaction energy at its product state (Eint0). The Eint0 values are similar among oxides unless a large charge-balancing cation is present and interacts with .OOH; consequently, HAE2 acts as an appropriate descriptor of the outer sphere dynamics. HAE2 also determines the thermodynamics of H2O dissociation on O-vacancies, which influence the inner and outer sphere rates by setting the relative coverage of * and H/OH*. These results, in turn, show that HAE2 is a complete descriptor of the reactivity and selectivity of oxides for O2 activation; the O-atoms in more reducible oxides (more negative HAE2) exhibit a greater preference for the inner sphere routes and for the formation of electrophilic OO* intermediates that mediate epoxidation and O-insertion reactions during catalytic redox cycles. Large charge-balancing cations locally modify Eint0 values that determine the outer sphere rates and thus can be used to alter the preference of O-atoms to either inner or outer sphere routes.
- S. Kwon, P. Deshlahra, and E. Iglesia. (2018) Dioxygen activation routes in Mars-van Krevelen redox cycles catalyzed by metal oxides, J. Cat., 364, 228 – 247.
Abstract and key graphics
Key graphics
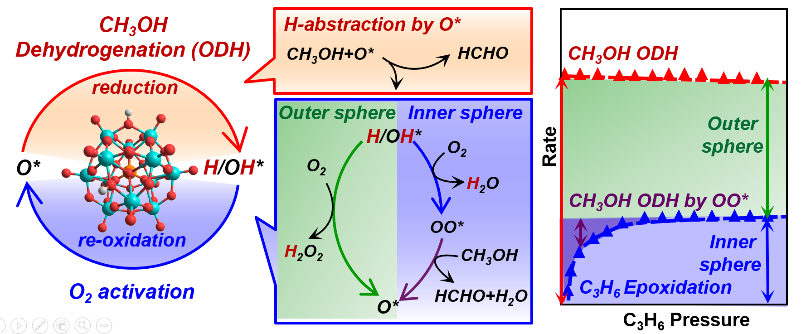
Highlights
- O2 activation involves inner and outer sphere routes during oxidative dehydrogenation
- Inner sphere routes form bound peroxo species that insert O-atoms into alkanols/alkenes
- Outer sphere routes form H2O2(g), O-atom shuttles that oxidize non-vicinal reduced centers
- These routes allow re-oxidation of two non-vicinal 2e− reduced centers by a 4e− oxidant (O2)
- Kinetic, scavenging, and theoretical methods can assess the contributions of each route
Abstract
Catalytic redox cycles involve dioxygen activation via peroxo (OO∗) or H2O2 species, denoted as inner-sphere and outer-sphere routes respectively, for metal-oxo catalysts solvated by liquids. On solid oxides, O2 activation is typically more facile than the reduction part of redox cycles, making kinetic inquiries difficult at steady-state. These steps are examined here for oxidative alkanol dehydrogenation (ODH) by scavenging OO∗ species with C3H6 to form epoxides and by energies and barriers from density functional theory. Alkanols react with O-atoms (O∗) in oxides to form vicinal OH pairs that eliminate H2O to form OO∗ at O-vacancies formed or react with O2 to give H2O2. OO∗ reacts with alkanols to re-form O∗ via steps favored over OO∗ migrations, otherwise required to oxidize non-vicinal vacancies. C3H6 epoxidizes by reaction with OO∗ with rates that increase with C3H6 pressure, but reach constant values as all OO∗ species react with C3H6 at high C3H6/alkanol ratios. Asymptotic epoxidation/ODH rate ratios are smaller than unity, because outer-sphere routes that shuttle O-atoms via H2O2(g) are favored over endoergic vacancy formation required for inner-sphere routes. The relative contributions of these two routes are influenced by H2O, because vacancies, required to form OO∗, react with H2O to form OH pairs and H2O2. OO∗-mediated routes and epoxidation become favored at low coverages of reduced centers, prevalent for less reactive alkanols and lower alkanol/O2 ratios, because H2O2 then reacts preferentially with O∗ (forming OO∗), instead of vacancies (forming O∗/H2O). Such kinetic shunts between two routes compensate for lower barriers required to form H2O2 than OO∗. These re-oxidation routes prefer molecular donor (H2O2) or acceptor (alkanol) to perform stepwise two-electron oxidations by dioxygen, instead of kinetically demanding O-atom migrations. The quantitative descriptions, derived from theory and experiment on Mo-based polyoxometalate clusters with known structures, bring together the dioxygen chemistry in liquid-phase oxidations, including electro-catalysis and monooxygenase enzymes, and oxide surfaces into a common framework, while suggesting a practical process for epoxidation by kinetically coupling with ODH reaction.
- S. Kwon, P. Liao, P. C. Stair, and R. Q. Snurr (2016) Alkaline-earth metal-oxide overlayers on TiO2: application toward CO2 photoreduction, Catal Sci Technol., 6, 7885 – 7895.
Abstract and key graphics
Key graphics
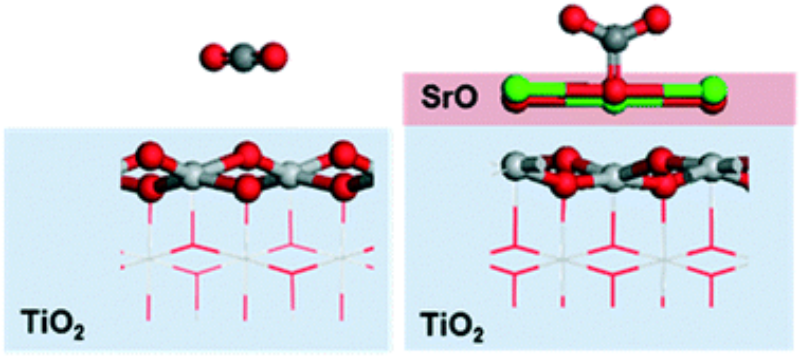
Abstract
Converting CO2 into valuable C1 products such as CO, methanol, and methane using photocatalysts is an attractive way to recycle atmospheric CO2 into fine chemicals and fuels. The most commonly studied photocatalyst, TiO2, however, suffers from poor initial adsorption of CO2. To overcome this problem, it has been proposed that a thin overlayer of a basic oxide might promote CO2 adsorption and thus improve the reactivity of TiO2 for photoreduction of CO2. In this work, we investigated CO2 adsorption on the (100) surfaces of a series of basic, alkaline-earth metal oxides (MgO, CaO, SrO, BaO). Using periodic density functional theory (DFT) calculations, we found that CO2 adsorption becomes significantly more favorable in the order MgO < CaO < SrO < BaO, and we attribute this order to the more suitable lattice parameter of BaO compared to MgO. To understand the effect of a thin layer of basic oxide on TiO2 for CO2 photoreduction, SrO on TiO2 was investigated as a model system. A dramatic improvement in CO2 adsorption and activation was observed on SrO/TiO2 compared to the bare TiO2, and dissociated water was found to be thermodynamically more favorable than intact water on the SrO/TiO2 surface. A possible reaction route for the photocatalytic reduction of CO2 to CO on the bare and SrO-modified TiO2 surfaces was further investigated. Although the reaction is slightly more favorable on the TiO2 surface than on the 0.5 ML SrO-covered TiO2, the SrO half layer helps activate CO2 and favors desorption of CO, which are challenging steps for CO2 reduction on pure TiO2. Therefore, our results suggest that <1 ML SrO overlayer might be a promising candidate for further experimental exploration.
- S. Kwon, N. M. Schweitzer, S. Y. Park, P. C. Stair, and R. Q. Snurr (2015) A kinetic study of vapor- phase cyclohexene epoxidation by H2O2 over mesoporous TS-1, J. Cat., 323, 117 - 115.
Abstract and key graphics
Key graphics
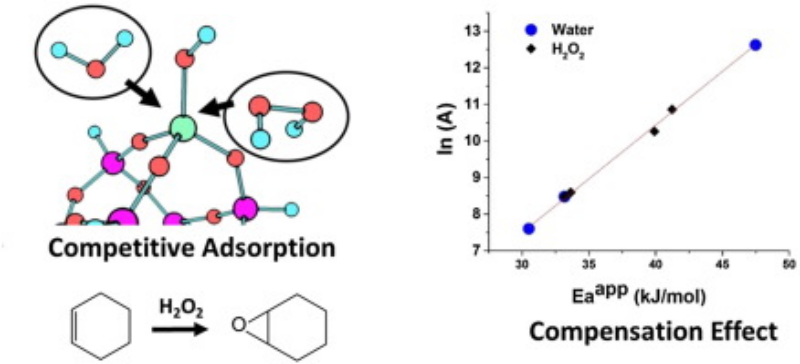
Highlights
- Vapor-phase cyclohexene epoxidation was performed over mesoporous TS-1 using H2O2
- The gas-phase production of cyclohexene epoxide was very stable with high selectivity
- Detailed kinetic studies were performed on gas-phase alkene epoxidation
- A compensation effect was observed with varied partial pressure of water or H2O2
- We report a kinetic model to understand the mechanism and the compensation effect
Abstract
A kinetic analysis of gas-phase cyclohexene epoxidation by H2O2 over mesoporous TS-1 was performed. The production of cyclohexene oxide was very stable with high selectivity. Based on the kinetic analysis, the gas-phase mechanism is proposed to be similar to that of the liquid-phase reaction: an Eley–Rideal type mechanism, in which the reaction between a Ti–OOH intermediate and the physisorbed alkene is the rate-determining step. When the partial pressure of water or H2O2 was varied, a compensation effect was observed. Based on the kinetic model, the compensation effect is attributed to variations in the surface coverage of intermediates, specifically the competitive adsorption of water and H2O2 at the Ti active sites. A meaningful activation energy can only be obtained at high surface coverages of H2O2 and was determined to be 40 ± 2 kJ/mol.
- Tuci, G., G. Giambastiani, S. Kwon., P. C. Stair, R. Q. Snurr, and A. Rossin (2014) Chiral Co(II) metal– organic framework in the heterogeneous catalytic oxidation of alkenes under aerobic and anaerobic Conditions, ACS Catal., 4, 1032 – 1039.
Abstract and key graphics
Key graphics
Abstract
The chiral Co(II) MOF [Co(L-RR)(H2O)·H2O]∞ [1; L-RR = (R,R)-thiazolidine-2,4-dicarboxylate] has been exploited in the catalytic oxidation of different alkenes (cyclohexene, (Z)-cyclooctene, 1-octene) using either tert-butyl hydroperoxide (tBuOOH) or molecular oxygen (O2) as oxidants. Different chemoselectivities are observed, both substrate- and oxidant-dependent. A moderate enantioselectivity is also obtained in the case of prochiral precursors, revealing the chiral induction ability of the optically pure metal environment. The interaction of O2 with the exposed metal sites in 1 (after material preactivation and consequent removal of the coordinated aquo ligand) has been studied through TPD-MS analysis combined with DFT calculations, with the aim of probing effective oxygen uptake by the heterogeneous catalyst and unraveling the nature of the active species in the catalytic oxidation process under aerobic conditions. Theoretical results indicate the presence of an η1-superoxo species at the cobalt center, with concomitant Co(II) ↔ Co(III) oxidation. Finally, the experimental estimation of the O2 adsorption enthalpy is found to be in good agreement with the calculated binding energy.
- Mondloch, J. E., W. Bury, D. Fairen-jimenez, S. Kwon, E. J. Demarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha, and J. T. Hupp (2013) Vapor-phase metalation by atomic layer deposition in a metal−organic framework, J. Am. Chem. Soc., 135, 10294 - 10297 (Highlighted in Chemical & Engineering News).
Abstract and key graphics
Key graphics
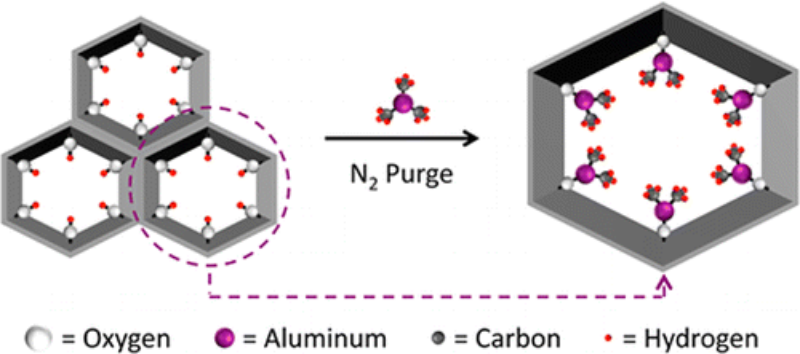
Abstract
Metal–organic frameworks (MOFs) have received attention for a myriad of potential applications including catalysis, gas storage, and gas separation. Coordinatively unsaturated metal ions often enable key functional behavior of these materials. Most commonly, MOFs have been metalated from the condensed phase (i.e., from solution). Here we introduce a new synthetic strategy capable of metallating MOFs from the gas phase: atomic layer deposition (ALD). Key to enabling metalation by ALD In MOFs (AIM) was the synthesis of NU-1000, a new, thermally stable, Zr-based MOF with spatially oriented −OH groups and large 1D mesopores and apertures.